6.19. The ORCA DOCKER: An Automated Docking Algorithm¶
The most important aspects of chemistry/physics do not occur with single molecules, but when they interact with each other. Now, given any two molecules, how to put them together in the best interacting “pose”? That is what we try to answer when using the ORCA DOCKER. Docking here refers to the process of taking two systems and putting them together in their best possible interaction.
6.19.1. Example 1: A Simple Water Dimer¶
Let us start with a very simple example. Given two water molecules, how to find the optimal dimer? With the DOCKER that is simple and can be done with:
!XTB
%DOCKER GUEST "water.xyz" END
* xyz 0 1
O -2.13487 2.63905 -0.01809
H -1.16698 2.61938 0.02397
H -2.41372 2.24598 0.82256
*
where the file water.xyz is a .xyz file which contains the same water structure, optionally with charge and multiplicity (in that order) on the comment line (the second line by default):
3
0 1
O -2.13487 2.63905 -0.01809
H -1.16698 2.61938 0.02397
H -2.41372 2.24598 0.82256
The molecule given on the regular ORCA input will be the HOST, and the GUEST is always given through an external file.
The output will start with:
***************
* ORCA Docker *
***************
Reading guests from file water.xyz
Number of structures read from file 1
Charge and multiplicity of guest from file
Docking approach independent
Docking level normal
Optimizing host .... -5.070544 Eh
Optimizing guest .... -5.070544 Eh
where it writes the name of the file with the GUEST structure, the number of structures read, some extra info and will optimize both host and guest (in this case they are the same), here by default using GFN2-XTB.
Note
If no multiplicity or charge are given, the GUEST is assumed to be neutral and closed-shell.
Note
The DOCKER right now is only working with the GFN-XTB and GFN-FF methods and the ALPB solvation model. It will be expanded later to others.
That is followed by some extra info that is explained in more details on its own detailed section (see More details on the ORCA DOCKER):
Starting Docker
---------------
Guest structure .... structure number 1
Guest charge and multiplicity .... (0 , 1)
Final charge and multiplicity .... (0 , 1)
PES used during evolution .... GFN2-XTB
Setting random seed .... done
Creating spatial grid
Grid Max Dimension 5.50 Angs
Angular Grid Step 32.73 degrees
Cartesian Grid Step 0.50 Angs
Points per Dimension 11 points
Initializing workers
Population Density 0.50 worker/Ang^2
Population Size 57
Evolving structures
Minimization Algorithm mutant particle swarm
Min, Max Iterations (3 , 10)
That is followed by the docking itself, which will stop after a few iterations:
Iter Emin avDE stdDE Time
(Eh) (kcal/mol) (kcal/mol) (min)
-------------------------------------------------------
1 -10.147462 2.756033 1.821981 0.03
2 -10.147462 2.121389 1.610208 0.03
3 -10.148583 2.313606 1.365227 0.03
4 -10.148583 1.846998 1.188680 0.02
5 -10.148583 1.587332 1.168207 0.02
No new minimum found after 3 (MinIter) steps.
The idea here is to collect as many local minima as possible, that is, collect a first guess for all possible modes of interaction between the different structures. We do this by allowing both structures to partially optimize, but it is important to say we will not look for multiple conformers of the host and guest here.
With all solutions collected, we will take a fraction of them and do a final full optimization:
Running final optimization
Maximum number of structures 7
Minimum energy difference 0.10 kcal/mol
Maximum RMSD 0.25 Angs
Optimization strategy regular
Coordinate system redundant 2022
Fixed host false
Struc Eopt Interaction Energy Time
(Eh) (kcal/mol) (min)
------------------------------------------------
1 -10.149006 -4.968378 0.01
2 -10.149005 -4.967965 0.01
3 -10.149007 -4.968825 0.01
4 -10.149007 -4.968641 0.01
5 -10.149007 -4.968743 0.01
6 -10.149006 -4.968116 0.01
7 -10.149007 -4.968678 0.01
And as you can see, we also automatically print the Interaction Energy, which is simple an energy difference between the final complex, host and guest. The final best structure with lowest interaction energy is then saved on the Basename.docker.xyz file. If needed, all other structures are saved on the Basename.docker.struc1.allopt.xyz, as written on the output:
All optimized structures saved to : Basename.docker.struc1.allopt.xyz
-------------------------------------
LOWEST INTERACTION ENERGY: -4.968825 kcal/mol (structure 3)
-------------------------------------
(...)
The lowest energy structure was 1, with energy -10.149007.
Docked structures saved to Basename.docker.xyz
Note
The name Basename.docker.struc1.allopt.xyz refers to struc1 because that is the first docked guest. Later that can be done with multiple guest and that is only a way to organize the outputs.
We are all set, the output can be visualized and it is, as expected:

Fig. 6.72 The final water dimer found using the GFN2-XTB PES.¶
6.19.2. Example 2: A Uracil Dimer¶
Now for a slightly more complex example, a uracil dimer:
! XTB PAL16
%DOCKER GUEST "uracil.xyz" END
*xyz 0 1
N -0.2707028 0.7632994 1.0276159
H -0.5957915 1.3097757 1.8163465
C -0.3386212 1.3810817 -0.2276640
O -0.7270425 2.5346295 -0.3329857
N 0.3638189 -1.2896563 0.1949192
H 0.0796815 0.9143946 -2.3190044
C 0.3781329 -0.7736192 -1.0714063
H 0.6499130 -1.4675080 -1.8526542
C 0.0669084 0.5154897 -1.3194961
H 0.4818502 -2.2779688 0.3498201
C -0.0016589 -0.5616540 1.3117092
O -0.0864879 -1.0482643 2.4227999
*
where the uracil.xyz is a simple repetition of the structure, as with the water before.
In this case the output is more diverse, and in fact many different poses appear as candidates for the final optimization:
Struc Eopt Interaction Energy Time
(Eh) (kcal/mol) (min)
------------------------------------------------
1 -49.248577 -11.723457 0.08
2 -49.250442 -12.893758 0.08
3 -49.245624 -9.870339 0.03
4 -49.252991 -14.493130 0.06
5 -49.248470 -11.656256 0.05
6 -49.259335 -18.474228 0.05
7 -49.259269 -18.432902 0.08
8 -49.254913 -15.699019 0.03
9 -49.254927 -15.708244 0.03
10 -49.241672 -7.390198 0.02
11 -49.246534 -10.441269 0.03
and structure number 6 is found to be the one with lowest interaction energy:
-------------------------------------
LOWEST INTERACTION ENERGY: -18.474228 kcal/mol (structure 6)
-------------------------------------
Here is a scheme with the structures found and their relative energies:
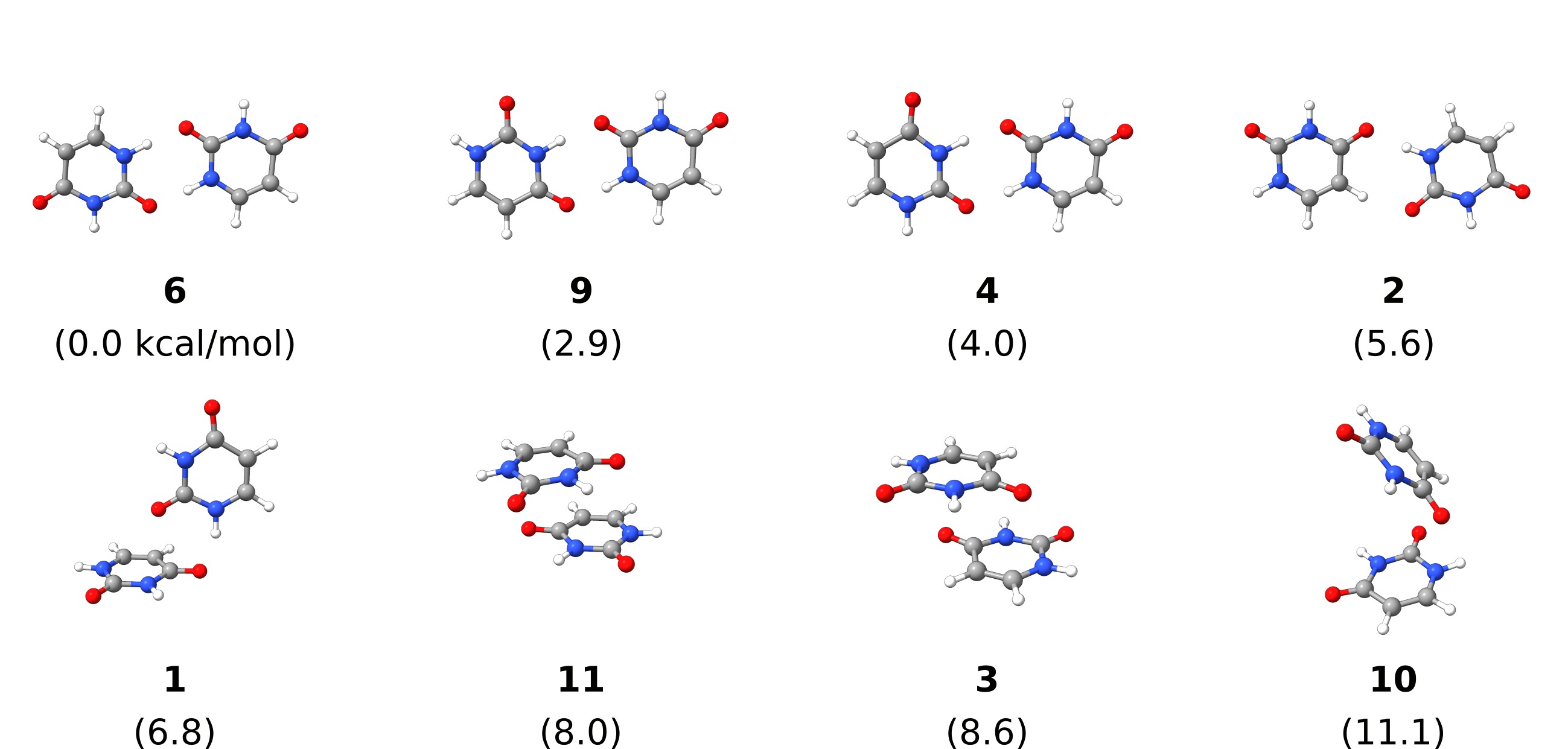
Fig. 6.73 Uracil dimer structures generated by DOCKER (duplicates removed) with relative energies in kcal/mol.¶
Note
There might be duplicated results after the final optimization, these are currently not automatically removed. Here they were manually removed.
Important
The PAL16 flag means XTB will run in parallel, but the ORCA DOCKER could be parallelized in a much more efficient way by paralleizing over the workers. That will be done for the next versions and it will be significantly more efficient.
6.19.3. Example 3: Adding Multiple Copies of a Guest¶
Suppose you want to add multiple copies of the same guest, for example three water molecules on top of the uracil one after the other. That can be simply done with:
! XTB PAL16
%DOCKER
GUEST "water.xyz"
NREPEATGUEST 3
END
*xyz 0 1
N -0.2707028 0.7632994 1.0276159
H -0.5957915 1.3097757 1.8163465
C -0.3386212 1.3810817 -0.2276640
O -0.7270425 2.5346295 -0.3329857
N 0.3638189 -1.2896563 0.1949192
H 0.0796815 0.9143946 -2.3190044
C 0.3781329 -0.7736192 -1.0714063
H 0.6499130 -1.4675080 -1.8526542
C 0.0669084 0.5154897 -1.3194961
H 0.4818502 -2.2779688 0.3498201
C -0.0016589 -0.5616540 1.3117092
O -0.0864879 -1.0482643 2.4227999
*
and the guests on water.xyz will be added on top of the previous best complex three times. Now, there will be files with names Basename.docker.struc1.allopt.xyz, Basename.docker.struc2.allopt.xyz and Basename.docker.struc3.allopt.xyz, one for each step. Still, the same final Basename.docker.xyz file and now a Basename.docker.build.xyz is also printed, with the best result after each iteration.
That’s how the results look like, from the Basename.docker.xyz:
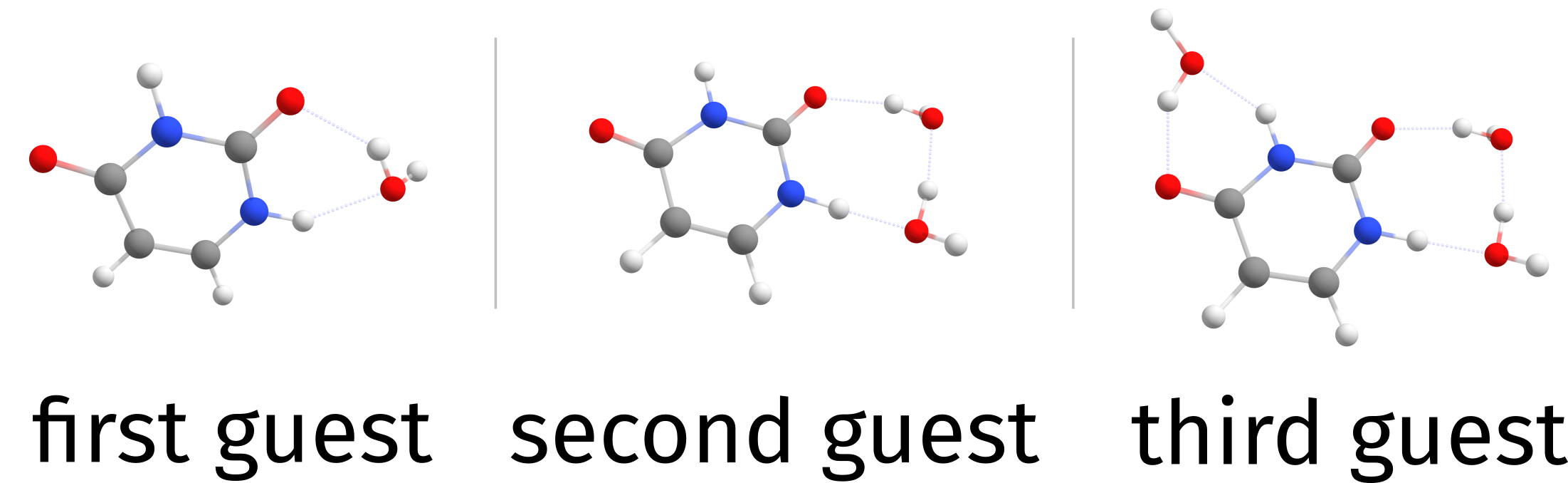
Fig. 6.74 Cumulative docking of three guests¶
Note
By default the HOST is always optimized. It can be changed with %DOCKER FIXHOST TRUE END.
6.19.4. Example 4: Find the Best Guest¶
Another common case would be: given a list of guests - or conformers of the same guest (see GOAT: global geometry optimization and ensemble generator) - one might want to know what is the “best guest”, that is the one with the lowest interaction energy.
In order to do that, simply pass a multixyz file and the DOCKER will try to dock all structures from that file, one by one:
! XTB
%DOCKER GUEST "uracil_water.xyz" END
*xyz 0 1
N -0.2707028 0.7632994 1.0276159
H -0.5957915 1.3097757 1.8163465
C -0.3386212 1.3810817 -0.2276640
O -0.7270425 2.5346295 -0.3329857
N 0.3638189 -1.2896563 0.1949192
H 0.0796815 0.9143946 -2.3190044
C 0.3781329 -0.7736192 -1.0714063
H 0.6499130 -1.4675080 -1.8526542
C 0.0669084 0.5154897 -1.3194961
H 0.4818502 -2.2779688 0.3498201
C -0.0016589 -0.5616540 1.3117092
O -0.0864879 -1.0482643 2.4227999
*
Here the file uracil_water.xyz looks like:
3
0 1
O -2.13487 2.63905 -0.01809
H -1.16698 2.61938 0.02397
H -2.41372 2.24598 0.82256
12
0 1
N -0.2707028 0.7632994 1.0276159
H -0.5957915 1.3097757 1.8163465
C -0.3386212 1.3810817 -0.2276640
O -0.7270425 2.5346295 -0.3329857
N 0.3638189 -1.2896563 0.1949192
H 0.0796815 0.9143946 -2.3190044
C 0.3781329 -0.7736192 -1.0714063
H 0.6499130 -1.4675080 -1.8526542
C 0.0669084 0.5154897 -1.3194961
H 0.4818502 -2.2779688 0.3498201
C -0.0016589 -0.5616540 1.3117092
O -0.0864879 -1.0482643 2.4227999
with a water followed by an uracil molecule. First, the water will be added, then the uracil, but both separately. The initial output is a bit different:
***************
* ORCA Docker *
***************
Reading guests from file uracil_water.xyz
Number of structures read from file 2
Charge and multiplicity of guests from file
Docking approach independent
Docking level normal
with now two structures being read from file, and the Docking approach is labeled as independent, meaning each structure will be docked independently of each other.
After everything, the output is:
-------------------------------------
LOWEST INTERACTION ENERGY: -18.482854 kcal/mol (structure 6)
-------------------------------------
Total time for docking: 4.84 minutes
The lowest energy structure was 2, with energy -49.259349.
Docked structures saved to Basename.docker.xyz
and one can see that the lowest interaction energy was that of structure 2 (the uracil), meaning it interacts strongly with the HOST than the water molecule given. Now the file Basename.docker.xyz will contain all final structures, ordered by interaction energy.
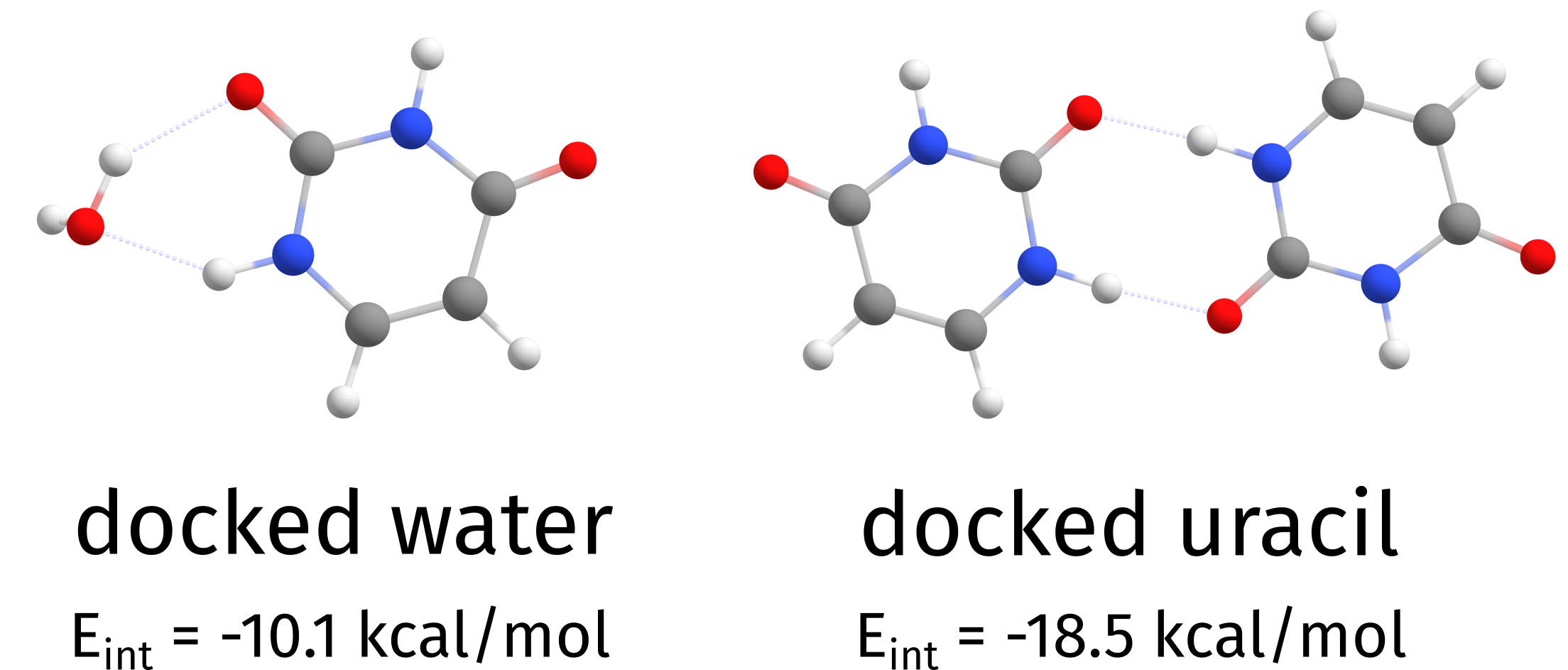
Fig. 6.75 Independent docking of water and uracil on top of an uracil molecule¶
Note
By default, the docking approach uses a fixed random seed and should always give the same result on the same machine. To make it always completely random add %DOCKER RANDOMSEED TRUE END.
Note
In order to use the faster GFN-FF instead of GFN2-XTB, use !DOCK(GFNFF). For a quicker (and less accurate) docking, use !QUICKDOCK.
Note
To try multiple conformers of the GUEST, the ensemble file printed by GOAT Basename.finalensemble.xyz can be directly given here and the whole ensemble will be tested against a give HOST.
A detailed description of the other options can be found on More details on the ORCA DOCKER
6.19.5. Reduced Keyword List¶
!QUICKDOCK # simple keyord to set DOCKLEVEL QUICK
!NORMALDOCK # simple keyord to set DOCKLEVEL NORMAL
!COMPLETEDOCK # simple keyord to set DOCKLEVEL COMPLETE
!DOCK(GFN-FF) # simple keyord to set EVPES GFNFF
!DOCK(GFN0-XTB) # simple keyord to set EVPES GFN0XTB
!DOCK(GFN1-XTB) # simple keyord to set EVPES GFN1XTB
!DOCK(GFN2-XTB) # simple keyord to set EVPES GFN2XTB
%DOCKER
#
# general options
#
GUEST "filename.xyz" # an .xyz file (can be multistructure), from where
# the guest(s) will be read. can contain different
# charges and multiplicities for each guests on the
# comment line. will only be read if exactly two
# integer numbers are given, otherwise ignored.
DOCKLEVEL SCREENING # defines a general strategy for docking.
# will alter things like that population density
NORMAL # and final number of optimized structrures.
COMPLETE # default is NORMAL.
NREPEATGUEST 1 # number of times to repeat the content of the "GUEST" file
CUMULATIVE TRUE # add the contents of the "GUEST" file one on
# top of each other?
# default is FALSE, meaning each will be done independently.
FIXHOST TRUE # freeze coordinatef for the HOST during all steps?
# (default FALSE)
#
# evolution step
#
EVPES GFNFF # which PES to use **only** during the evolution step.
GFN0XTB # can be different from the final optimization.
GFN1XTB
GFN2XTB
#
# final optimization
#
NOPT 10 # a fixed number of structures to be optimized
NOOPT FALSE # do not optimize any structure at all? (default FALSE)